
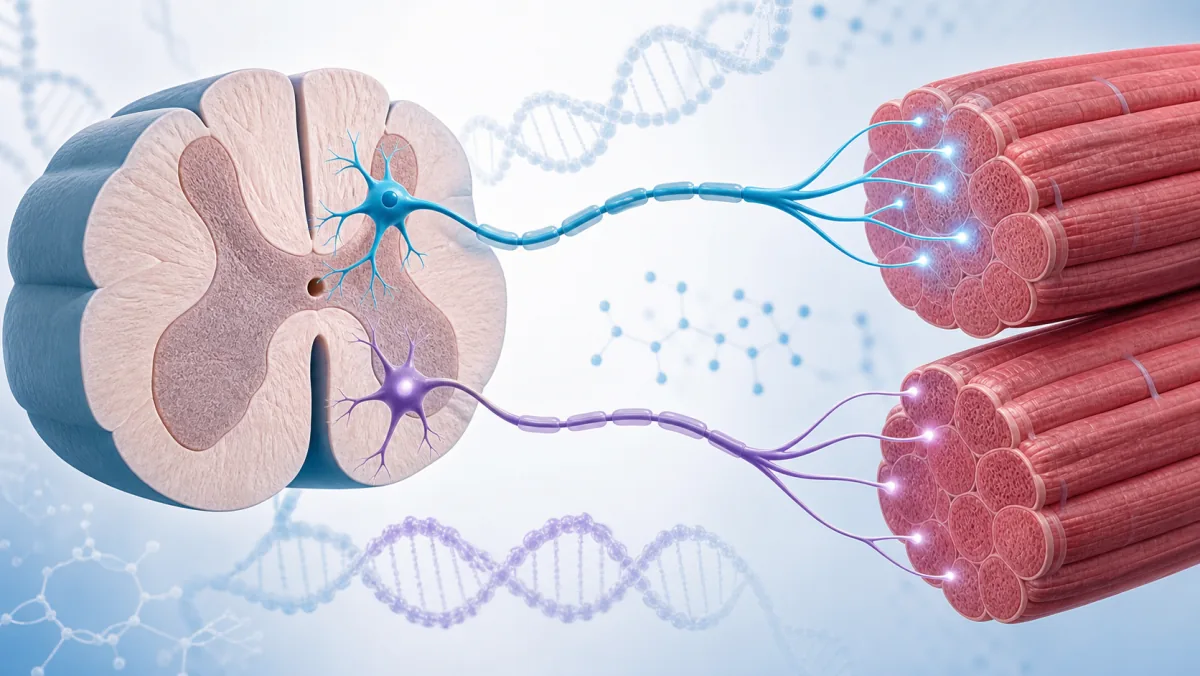
Svaki pokret koji naše telo pravi kontroliše dugačka, tanka struktura koja se naziva „kičmena moždina“. Kičmena moždina je deo našeg nervnog sistema i povezuje mozak sa donjim delom leđa. Sastoji se od motornih nervnih ćelija koje prenose signale između mozga i nerava širom tela, kontrolišući pokrete i osećaje poput bola.
Šta je i šta uzrokuje SMA?
SMA (Spinalna mišićna atrofija) je retka, progresivna i teška neuromišićna bolest. To je genetički poremećaj koji direktno napada motorne neurone u kičmenoj moždini, što dovodi do propadanja, atrofije i slabljenja mišića.
SMA nastaje usled promena u genima za spinalnu mišićnu atrofiju – SMN1 i SMN2, što dovodi do veoma niskog nivoa ili potpunog nedostatka SMN proteina (Survival Motor Neuron protein). SMN protein ima ključnu ulogu u brojnim ćelijskim procesima, uključujući funkciju i održavanje motornih neurona i slanju signal ćelijama.
Gen SMN1 je odgovoran za stvaranje dovoljne količine funkcionalnog SMN proteina. Otkrivene su brojne genetičke promene u SMN1 genu koje dovode do stvaranja nefunkcionalnog SMN proteina i razvoja SMA.
Gen SMN2, poznat je kao „rezervni gen“, proizvodi samo oko 10% SMN proteina u odnosu na SMN1 gen. Dok je SMN1 odgovoran za nastanak bolesti, SMN2 utiče na težinu simptoma SMA. Obično osoba ima jednu do dve kopije SMN2 gena, ali neki ljudi mogu imati i do osam kopija. Uopšteno, što osoba ima više kopija SMN2 gena, simptomi bolesti su blaži. Zbog uticaja na tok bolesti, SMN2 se često naziva „modifikator bolesti“ . U literature postoje i drugi geni koji utiču na modifikaciju bolesti.
Koliko je česta spinalna mišićna atrofija (SMA)?
Iako se SMA smatra retkom bolešću, pogađa približno 1 na 6.000 do 1 na 10.000 živorođene dece. Procene pokazuju da je širom sveta oko 1 od 50 osoba nosilac SMA mutacije.
Od uvođenja obaveznog neonatalnog skrininga nakon rođenja, testirano je više od 136.000 beba, a SMA povrđena kod oko 20 novorođenčadi.
Nosioci nemaju simptome bolesti jer poseduju jednu zdravu i jednu izmenjenu kopiju SMN gena. Međutim, pošto se SMA nasleđuje autosomno – recesivno, roditelji koji su oboje nosioci imaju 25% šanse da dobiju dete obolelo od SMA.
Zbog relativno visoke učestalosti nosilaca SMA mutacija, Američki koledž ginekologa i akušera (ACOG) preporučuje da svim ženama koje planiraju trudnoću ili su već trudne bude ponuđeno testiranje na nosilaštvo za SMA.
Veoma mali procenat SMA slučajeva nastaje „de novo“ (spontano) prvi put, bez toga da su roditelji nosioci mutacije.
Više o proveri rizika za nasledna oboljenja dostupno je na stranici nasledne bolesti.
Koji su simptomi i tipovi SMA?
Simptomi SMA značajno variraju, ali su najčešći mišićna slabost i hipotonija (smanjen tonus mišića). Danas se zna da SMA deli na 5 tipova, prema težini simptoma, uzrastu pojave bolesti kao i broju kopija SMN2 gena.
SMA tip 0
SMA tip 0 predstavlja prenatalni oblik bolesti i najređi je, ali i najteži oblik, koji pogađa oko 1% pacijenata. Obolela deca imaju samo jednu kopiju SMN2 gena. Simptomi mogu početi još tokom trudnoće, kroz smanjene ili odsutne pokrete fetusa. Po rođenju prisutni su hipotonija, izražena mišićna slabost, urođene srčane mane i nekroza kože. Bez lečenja, smrt može nastupiti u prvim nedeljama života.
SMA tip 1
SMA tip 1 je najčešći oblik bolesti i javlja se u oko 60% slučajeva. Pacijenti obično imaju 2–3 kopije SMN2 gena. Simptomi se javljaju između rođenja i 6. meseca života.
Obolele bebe, poznate kao „non-sitters“, nemaju kontrolu glave i ne mogu samostalno da sede zbog izražene hipotonije. To dalje dovodi do problema sa disanjem, hranjenjem i gutanjem, povećavajući rizik od pothranjenosti i plućnih infekcija. Može se javiti i karakterističan „žablji položaj“ nogu pri ležanju.
Bez terapije, bolest često ima fatalan ishod do druge godine života.
SMA tip 2
Pacijenti sa SMA tipom 2 obično imaju 3 kopije SMN2 gena, a simptomi se javljaju između 6. i 18. meseca života.
Nazivaju se „sitters“ jer mogu samostalno da sede, ali ne mogu da stoje ili hodaju bez pomoći. Simptomi su blaži nego kod tipa 1 i uključuju otežano gutanje, slabost ruku i nogu, tremor prstiju i šaka, deformitete kičme poput skolioze, kao i probleme sa disanjem koji mogu dovesti do infekcija pluća. Većina osoba sa SMA tipom 2 može živeti duže od 25 godina čak i bez terapije.
SMA tip 3
Pacijenti sa SMA tipom 3 imaju 3–4 kopije SMN2 gena. Simptomi se mogu pojaviti između 18 meseci i 3 godine života ili između 3. i 30. godine.
Ovi pacijenti se nazivaju „walkers“ jer mogu samostalno da hodaju i stoje. Najčešće nemaju poteškoće sa gutanjem i hranjenjem, a uz odgovarajuću terapiju očekivani životni vek je normalan. Ipak, mogu imati probleme sa ravnotežom, abnormalan hod i postepeni gubitak sposobnosti hodanja.
SMA tip 4
SMA tip 4 je oblik bolesti koji se javlja kod odraslih i čini manje od 1% svih slučajeva SMA. Pacijenti imaju 4 do 8 kopija SMN2 gena.
Simptomi se pojavljuju nakon 30. godine života i mogu uključivati tremor, trzaje mišića i slabost nogu. Osobe sa ovim tipom SMA imaju normalan životni vek i uglavnom zadržavaju sposobnost samostalnog hodanja.
Pored SMA izazvane promenama u SMN1 genu, postoje i drugi oblici spinalne mišićne atrofije uzrokovani mutacijama u drugim genima.
Kako se postavlja dijagnoza SMA?
Na SMA se najčešće posumnja kada postoji porodična istorija bolesti, simptomi hipotonije (smanjene mišićne snage ili odsustva motorne snage) i kašnjenje u dostizanju razvojnih motoričkih prekretnica u ranom detinjstvu. Pošto simptomi SMA mogu biti veoma slični drugim neuromišićnim oboljenjima dijagnoza se kasnije postavlja i zato se pored kliničkog pregleda preporučuje se genetičko testiranje gena SMN1 i SMN2 kao dijagnostička metoda za potvrdu SMA, procenu težine bolesti i određivanje prognoze.
Rana dijagnoza SMA zašto je važna?
Rano prepoznavanje i dijagnostikovanje SMA od ključnog je značaja, ne samo zato što je oštećenje motornih neurona nepovratno, već i zato što omogućava pravovremeni pristup kliničkim studijama, terapijama koje mogu spasiti život i multidisciplinarnoj nezi koja pomaže deci da dostignu motoričke razvojne prekretnice.
Iz tog razloga SMA se sve češće uključuje u nacionalne programe neonatalnog skrininga širom sveta što je od nedavno slučaj i sa našom zemljom – tj moguće je testiranje dece nakon rođenja.

Da li se testiranje može sprovesti i pre rođenja dece?
Planiranje trudnoće danas više ne počinje tek kada žena zatrudni. Savremena medicina omogućava da se veliki broj genetskih rizika prepozna pre začeća ili u ranoj trudnoći, što budućim roditeljima daje nešto izuzetno važno — vreme i informaciju da donesu odluke na osnovu činjenica.
Genetičko testiranje nije „obavezno“, ali je sve više deo odgovorne pripreme za trudnoću, posebno kod parova koji žele da smanje rizik od naslednih bolesti kod deteta.
Genetika pre rođenja: šta zapravo znači „testiranje pre trudnoće“?
Genetičko testiranje može se uraditi:
- Pre trudnoće (prekonceptivno testiranje)
- Tokom trudnoće (prenatalno testiranje)
- Posle rođenja (neonatalni skrining i dijagnostika)
Genetičko testiranje pre trudnoće (prekonceptivno)
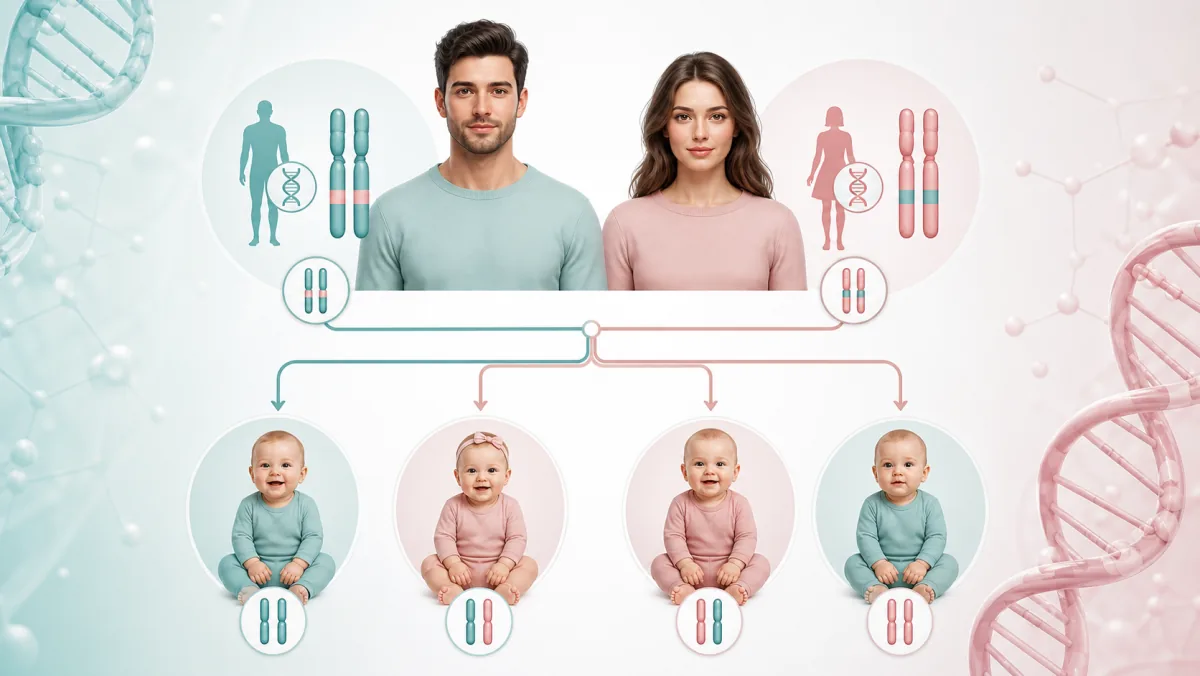
Ovo je jedna od najvažnijih, ali i najčešće preskočenih faza. Cilj prekonceptivnog testiranja je da se utvrdi da li su budući roditelji nosioci istih recesivnih genetskih mutacija.
To znači:
roditelji su najčešće potpuno zdravi nosioci i bez simptoma, ali ako oba roditelja nose istu mutaciju, postoji:
25% šanse da dete bude obolelo
50% šanse da bude nosilac
25% šanse da bude potpuno bez mutacije
Kada ovo znamo jedino par koji zna svoj genetski status može:
- planirati trudnoću uz adekvatno medicinsko savetovanje
- uraditi dodatne genetike testove u trudnoći za proveru bebe
- razmotriti IVF sa genetičkom selekcijom embriona (PGT-M)
Genetičko testiranje tokom trudnoće (prenatalno testiranje)
Ako trudnoća već postoji, genetička se može raditi na više načina:
- U ranoj trudnoći jos uvek se mogu uraditi skrining testovi koji podrazumevaju ispitivanje zdravih osoba, mladih bračnih parova Na taj način dobijamo informaciju da li su ti parovi zdravi nosioci bez simptoma ili su ne nose genetičku promenu.
Na osnovu ovog statusa dalje se određuju potrebna ispitivanja pre rođenja.
Uz PREGNAnipt neinvazivni prenatalni test dostupno je testiranje i ispitivanje statusa nosilaca budućih roditelja tokom trudnoće na neinvazivan i bezbedan način.
Više o neinvazivnom prenatalnom testiranju možete pronaći na stranici prenatalni test.

- Dijagnostičke metode su metode koje takođe radimo u toku trudnoće. One nose i mali, ali realan medicinski rizik, pa se rade samo kada postoji indikacija
U toku trudnoće radi se u situaciji kada oba biološka roditelja već imaju dokazan status nosioca promene za spinalnu miđićnu atrofiju – SMA. Tada se uzima uzorak plodove vode (amniona) koji se analizira i na osnovu analize amnion dobijamo informaciju da li je beba obolela, nosilac ili ne nosi gentičku promenu.
Genetičko testiranje posle rođenja
U mnogim zemljama, uključujući i Srbiju, postoji neonatalni skrining — testiranje novorođenčadi na određene metaboličke i genetske bolesti nakon što se dete rodi. Ovakva vrsta skrininga radi se u porodilištu.
Da li svi parovi treba da rade genetičko testiranje?
Ne postoji univerzalni odgovor, ali testiranje se posebno savetuje ako:
- postoji porodična istorija genetskih bolesti
- partneri potiču iz iste etničke grupe sa većom učestalošću određenih mutacija
- postoji raniji pobačaj ili neobjašnjiv gubitak trudnoće
- par želi planiranu i informisanu trudnoću
- trudnoća se planira u kasnijim godinama
Međutim, sve više parova bez ikakvih simptoma ili istorije bolesti bira preventivno testiranje, upravo zato što su nosioci često potpuno zdravi.
Genetičko testiranje pre i tokom trudnoće ne menja samo medicinski pristup — menja i način na koji roditelji doživljavaju trudnoću.
Da li postoji terapija za SMA?
Više od jednog veka SMA se smatrala neizlečivom bolešću. To se promenilo nakon otkrivanja funkcije SMN gena, što je omogućilo razvoj više terapijskih pristupa usmerenih direktno na uzrok bolesti.
Većina terapija ima cilj da poveća stvaranje SMN proteina — ili ponovnim aktiviranjem gena SMN1, ili modifikovanjem gena SMN2 kako bi proizvodio više funkcionalnog SMN proteina.
Za svaku terapiju postoje različiti kriterijumi, poput uzrasta pacijenta, telesne težine ili broja kopija SMN2 gena, ali najvažniji faktor za uspeh terapije i bolji kvalitet života jeste vreme započinjanja lečenja.
Kada se terapija započne pre pojave simptoma ili neposredno nakon njihovog nastanka, može doći do značajnog očuvanja motornih neurona i boljeg razvoja motoričkih funkcija.
Nusinersen (Spinraza)
Spinraza deluje na gen SMN2 povećavajući stvaranje funkcionalnog SMN proteina. Pokazala je značajnu efikasnost u poboljšanju motornih sposobnosti, respiratorne funkcije i preživljavanja kod odojčadi, dece i odraslih.
Onasemnogen abeparvovek (Zolgensma)
Zolgensma je prva odobrena genska terapija za neuromišićna oboljenja. FDA ju je odobrila 2019. godine, a EMA 2022. godine.
Lek se daje jednokratno intravenskim putem i sadrži funkcionalnu kopiju gena SMN1, čime omogućava stvaranje potrebnog SMN proteina.
Rana primena kod dece mlađe od dve godine pokazala je poboljšanje kontrole glave, sposobnosti samostalnog sedenja i razvoja govora.
Risdiplam (Evrysdi)
Kao i Spinraza, Evrysdi povećava proizvodnju funkcionalnog SMN proteina, što dovodi do poboljšanja sposobnosti sedenja, smanjenja problema sa gutanjem i unapređenja motoričkih funkcija i razvojnih prekretnica uz visok nivo bezbednosti i efikasnosti.
Pored postojećih terapija, u toku su brojna klinička ispitivanja novih pristupa lečenju.

PregnaTest kontakt
Telefon: +381658210919
Email: [email protected]